Molekulardynamische Bestimmung von Schmierstoff-Viskositäten
Dr. Kerstin Falk
Zur optimalen Auslegung geschmierter tribologischer Kontakte ist die Kenntnis der Schmierstoffeigenschaften unter den im belasteten Kontakt vorherrschenden Bedingungen notwendig. Empirische Formeln zur Vorhersage der Viskosität, wie Barus oder Roelands, werden üblicherweise an experimentelle Werte im Normaldruck-Bereich angefittet und zu höheren Drücken extrapoliert. Für die in Reibkontakten lokal auftretenden extremen Pressungen (im GPa Bereich) können sie allerdings versagen.
Atomistische Molekulardynamik Simulationen erlauben die numerische Berechnung von Schmierstoff-eigenschaften unter kontrollierten, nahezu beliebig einstellbaren, Druck- und Temperaturbedingungen. In diesem Projekt wurde dies zunächst für die Viskositätsberechnung verschiedener Modell-Schmierstoffe aus linearen und verzweigten Alkanen bei Drücken bis etwa 0.7 GPa demonstriert.
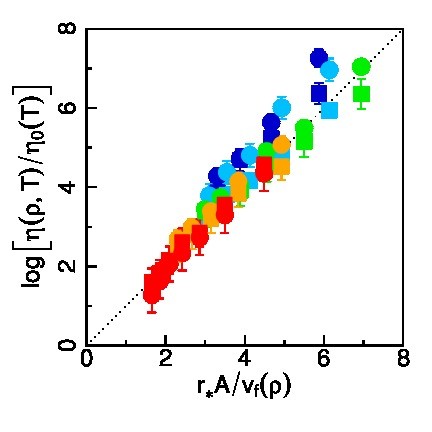
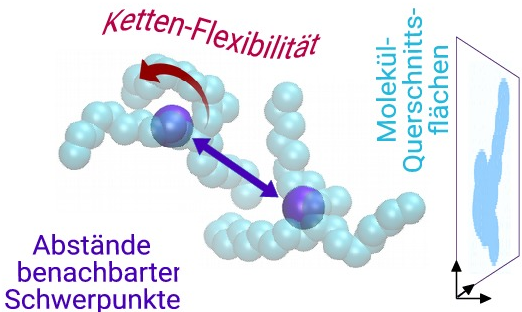
Über die explizite Berechnung der Viskosität hinaus, ermöglicht die atomistische Simulation auch eine mikroskopische Betrachtung der Schmierstoff Struktur und Dynamik. Mittels geeigneter statistischer Analysen konnte dadurch für Alkan-basierte Schmierstoffe ermittelt werden, welche molekularen Größen die Viskosität bestimmen. Daraus resultiert ein physikalisch motiviertes Viskositätsmodell, das nur von der Schmierstoff-Dichte und Temperatur, sowie von wenigen Molekül-Strukturgrößen abhängt. Diese Strukturgrößen sind der mittlere effektive Moleküldurchmesser, der mittlere Abstand zwischen benachbarten Molekülen, sowie die Flexibilität der Alkanketten. Diese drei Größen sind weitgehend unabhängig von der Dichte und können daher alle aus einer einzigen Molekulardynamik Simulation bei beliebiger Dichte -- beziehungsweise Druck, z.B. Normaldruck -- bestimmt werden. Zusammen mit einer akkuraten Zustandsgleichung für Druck, Temperatur und Dichte ergibt sich mit wenig Aufwand ein parameterfreies Viskositätsmodell, das auch als Konstitutivgesetz in Reynoldssolver implementiert werden kann.
Dieses physikalisch-basierte Viskositätsmodells soll in Zukunft gezielt adaptiert werden, um weitere anwendungsrelevante Bedingungen abbilden zu können. Dies beinhaltet als nächsten Schritt eine Erweiterung auf die rheologischen Eigenschaften in engen Spalten, die insbesondere auf Grund der Verwendung immer dünnflüssigerer Öle eine zunehmend wichtige Rolle spielen: Eine korrekte Beschreibung dieses Reibregimes mit nanoskaligen Schmierfilmdicken ist mit einer EHD-Betrachtung möglich, für die entsprechende Konstitutivgesetze auf der Nanoskala jedoch noch fehlen. Am IWM stehen atomistische Simulationen als ideales Werkzeug bereit, um die notwendigen Konstitutivgesetze aufstellen zu können.
Die bisherigen theoretischen Arbeiten zum Viskositätsmodell befinden sich aktuell bei der Zeitschrift Physical Review Letters unter Begutachtung.