Dr. Kerstin Falk
Zur optimalen Auslegung geschmierter tribologischer Kontakte ist die Kenntnis der Schmierstoffeigenschaften unter den im belasteten Kontakt vorherrschenden Bedingungen notwendig. Empirische Formeln zur Vorhersage der Viskosität, wie Barus oder Roelands, werden üblicherweise an experimentelle Werte im Normaldruck-Bereich angefittet und zu höheren Drücken extrapoliert. Für die in Reibkontakten lokal auftretenden extremen Pressungen (im GPa Bereich) können sie allerdings versagen. Ebenso fehlt bisher eine korrekte Beschreibung des Einflusses von Spalthöhe und Grenzflächeneigenschaften auf die Rheologie in engen Spalten, die auf Grund des Trends zu dünnflüssigeren Ölen immer wichtiger werden.
Um die Vorhersage von Schmierstoffeigenschaften zu verbessern, kommen am Fraunhofer IWM Molekulardynamik Simulationen zum Einsatz. Diese erlauben einerseits die numerische Berechnung von Schmierstoffeigenschaften unter kontrollierten tribologischen Bedingungen (z.B. Druck- und Temperatur, Scherraten und nanoskalige Spaltgröße). Andererseits ermöglicht die atomistische Simulation auch eine mikroskopische Betrachtung der Schmierstoffstruktur und -dynamik. Dadurch können Struktur-Eigenschaft Beziehungen identifiziert werden, die Materialgrößen wie etwa die Viskosität mit atomistischen Größen verknüpfen, und so physikalisch basierte Konstitutivgesetze für die makroskopische Modellierung liefern.
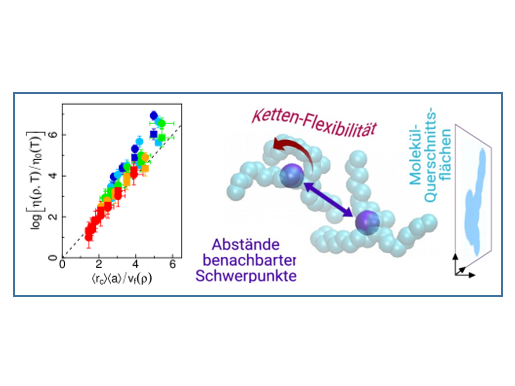
Ein mit diesem Ansatz erarbeitetes Hochdruck-Viskositätsmodell wurde nun in der Zeitschrift Physical Review Letters veröffentlicht [1]. In dieser Arbeit wurde für kohlenwasserstoffbasierte Schmierstoffe ermittelt, welche molekularen Größen die Viskosität bestimmen. Neben der Schmierstoffdichte und Temperatur, sind dies im Wesentlichen drei molekulare Strukturgrößen: der mittlere effektive Moleküldurchmesser, der mittlere Abstand zwischen benachbarten Molekülen, sowie die Flexibilität der Alkanketten (siehe Abb.1). Diese Größen sind weitgehend unabhängig von der Dichte und können daher alle aus einer einzigen Molekulardynamik Simulation bei beliebiger Dichte -- beziehungsweise Druck, z.B. Normaldruck -- bestimmt werden. Zusammen mit einer akkuraten Zustandsgleichung für Druck, Temperatur und Dichte ergibt sich mit wenig Aufwand ein parameterfreies Viskositätsmodell, das auch als Konstitutivgesetz in Reynoldssolver implementiert werden kann.
[1] https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.124.105501